
EDUCATIONAL SERIES
Content
The Physiology of Harm
A clinical and scientific diagnosis of alcohol’s damage to neurons, organs, and systemic health
Executive Summary
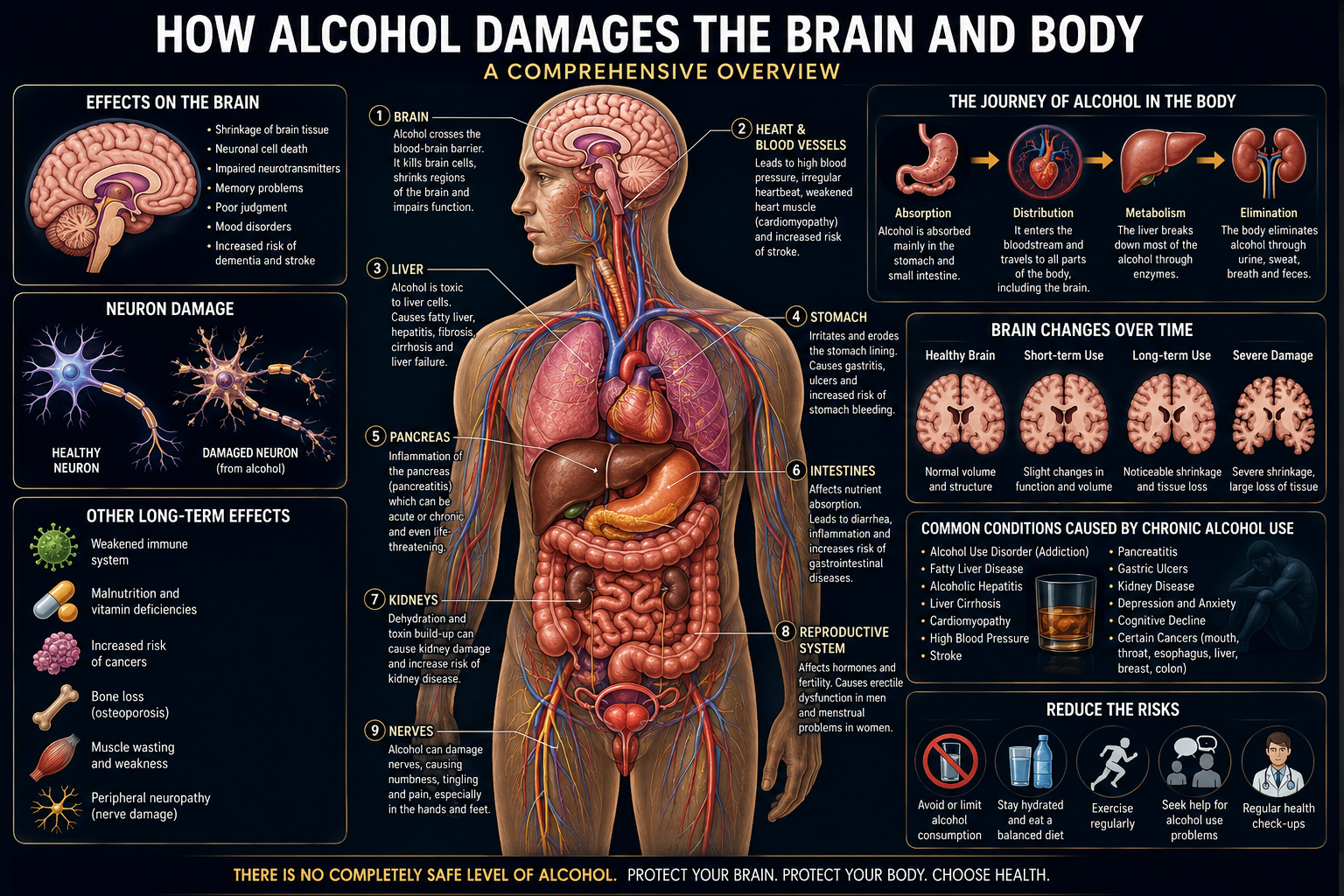
Ethanol (C₂H₅OH) is classified pharmacologically as a central nervous system depressant, yet its clinical footprint extends far beyond the brain. This article presents a systems-level diagnostic review of how chronic and acute alcohol exposure damages neurons and disrupts the structural and functional integrity of the liver, heart, pancreas, gastrointestinal tract, immune system, endocrine axis, musculoskeletal system, and reproductive organs.
The review is organised as a diagnostic reference: each organ system is examined through its mechanism of injury, the clinical and laboratory markers used to detect damage, and the staging of disease from early, reversible dysfunction to end-stage, irreversible failure.
KEY FINDINGThere is no organ system in the human body that is fully spared from ethanol’s toxic metabolites. The brain, liver, heart, pancreas, and immune system each accumulate measurable, dose-dependent damage, and several of these injuries — particularly neuronal and hepatic — follow a trajectory from reversible adaptation to permanent structural loss if exposure continues unchecked.
This document is intended as an educational and scientific reference. It does not constitute individual medical advice, and any person concerned about their own alcohol consumption or symptoms should consult a licensed physician or addiction medicine specialist. 📋 Copy Full Article Text
1. Introduction: Alcohol as a Systemic Toxin
Ethanol occupies a unique position in toxicology. Unlike most substances that damage a single target organ, ethanol and its primary metabolite, acetaldehyde, are directly cytotoxic across nearly every tissue type in the human body. This is a consequence of three properties: ethanol is small and freely water- and lipid-soluble, allowing it to cross essentially every biological membrane, including the blood-brain barrier and the placenta; it is metabolised primarily in the liver by enzymes that generate reactive, DNA-damaging intermediates; and its metabolism consumes and diverts cofactors, particularly NAD+ and thiamine, essential to normal cellular energy production throughout the body.
1.1 Scope of This Diagnostic Review
This article mirrors a systems-based clinical work-up: pharmacokinetics, then the central and peripheral nervous system, then the hepatic, cardiovascular, gastrointestinal, immune, endocrine, musculoskeletal, and reproductive systems. Each section identifies the dominant mechanism of injury, the diagnostic markers used to detect it, and where the damage sits on the spectrum from reversible to permanent.
1.2 Dose, Pattern, and Individual Variation
Severity is governed by total lifetime dose, pattern of consumption (chronic steady use versus episodic binge use), genetic variation in metabolic enzymes, sex, nutritional status, and co-existing conditions. Binge drinking favours acute neuronal excitotoxicity, arrhythmia, and acute pancreatitis; sustained chronic use favours cumulative neurodegeneration, cirrhosis, and cardiomyopathy.
CLINICAL NOTE — NO SAFE THRESHOLD FOR NEURONAL TISSUELarge-scale neuroimaging and epidemiological studies, including UK Biobank analyses, have found measurable reductions in grey matter volume associated with alcohol consumption even at levels previously considered “moderate,” with no clear lower threshold below which no structural brain effect is detectable.
2. Pharmacokinetics and Metabolism
2.1 Absorption and Distribution
Ethanol is absorbed rapidly by passive diffusion — about 20% across the gastric mucosa, the remainder across the proximal small intestine. Peak blood alcohol concentration is typically reached 30–90 minutes after ingestion on an empty stomach. Because ethanol distributes into total body water rather than fat, people with lower total body water reach higher blood concentrations from an equivalent dose.
2.2 The Oxidative Pathway
Over 90% of ingested ethanol is metabolised in the liver via alcohol dehydrogenase (ADH), producing acetaldehyde, then aldehyde dehydrogenase 2 (ALDH2), producing acetate. Acetaldehyde is a reactive electrophile that forms covalent adducts with DNA and proteins, and is classified by IARC as a Group 1 human carcinogen in the context of alcohol consumption. Variant ALDH2 alleles, common in people of East Asian descent, slow acetaldehyde clearance and raise oesophageal cancer risk substantially even at modest intake.
2.3 The Microsomal Pathway
With chronic heavy use, CYP2E1 (part of the microsomal ethanol-oxidising system) is upregulated, generating reactive oxygen species and contributing to oxidative liver injury, while also accelerating metabolism of many prescription drugs.
2.4 Cofactor Depletion: NAD+ and Thiamine
Ethanol metabolism consumes NAD+, shifting the liver’s redox state toward fat accumulation. Ethanol also impairs thiamine (B1) absorption and activation — a deficiency directly responsible for some of the most severe neurological complications of alcohol use (Section 5).
| Metabolic Step | Enzyme | Product | Toxic Consequence |
|---|---|---|---|
| Step 1 oxidation | ADH | Acetaldehyde | Reactive, DNA-adducting intermediate |
| Step 2 oxidation | ALDH2 | Acetate | Genetic variants slow clearance, raise cancer risk |
| Inducible pathway | CYP2E1 | Acetaldehyde + ROS | Oxidative stress, drug interactions |
| Cofactor cycling | NAD+ → NADH | Altered redox ratio | Suppressed fat oxidation, steatosis |
3. Neurological Damage — Mechanisms
The brain is disproportionately vulnerable because neurons are post-mitotic, long-lived cells that cannot be replaced in most regions once lost, and because ethanol interacts directly with the two principal neurotransmitter systems governing neuronal excitability.
3.1 Excitotoxicity and the GABA–Glutamate Imbalance
Ethanol acutely potentiates GABA-A receptors (inhibitory) and inhibits NMDA glutamate receptors (excitatory). With repeated exposure, neurons compensate by down-regulating GABA-A sensitivity and up-regulating NMDA receptor density. Abrupt withdrawal unmasks this adaptation, producing unopposed glutamate excitation, a calcium surge, and excitotoxic cell death — underlying withdrawal seizures and the “kindling” effect of repeated heavy-drinking-withdrawal cycles.
3.2 Oxidative Stress and Mitochondrial Dysfunction
Acetaldehyde and CYP2E1-derived reactive oxygen species overwhelm the brain’s limited antioxidant capacity, damaging neuronal mitochondrial DNA and membranes and impairing ATP production in cells with exceptionally high metabolic demand.
3.3 Neuroinflammation
Chronic exposure activates microglia toward a pro-inflammatory state. Ethanol also increases gut permeability, allowing bacterial endotoxin to enter circulation and cross into the brain, further activating microglia — a gut–brain inflammatory axis now recognised as a central driver of alcohol-related neurodegeneration.
3.4 Epigenetic and Neurogenesis Effects
Chronic exposure alters DNA methylation and histone acetylation in the hippocampus and prefrontal cortex, and suppresses adult neurogenesis in the hippocampal dentate gyrus — paralleling the memory deficits seen in chronic heavy drinkers.
MECHANISM SUMMARYExcitotoxicity, oxidative/mitochondrial injury, chronic neuroinflammation, and suppressed neurogenesis operate simultaneously and reinforce one another — why alcohol-related brain injury is typically worse than any single mechanism predicts.
4. Neurological Damage — Structural and Functional Consequences
4.1 Grey Matter Loss and Global Brain Volume
Chronic heavy drinkers show reduced whole-brain grey matter volume, most pronounced in the frontal lobes. The prefrontal cortex — responsible for executive function and impulse control — is particularly vulnerable, creating a self-reinforcing pattern: the region most damaged is also most responsible for regulating further intake.
4.2 The Cerebellum
The anterior superior vermis is among the most consistently affected structures. Purkinje cell loss produces the characteristic wide-based, unsteady gait and limb incoordination of alcoholic cerebellar degeneration — and is largely irreversible.
4.3 White Matter Integrity
Diffusion tensor imaging shows reduced white matter integrity in the corpus callosum and frontal tracts, reflecting myelin damage that slows processing speed and impairs inter-hemispheric communication.
4.4 The Hippocampus and Memory
Hippocampal volume reduction correlates with impaired formation of new long-term memories, compounded by suppressed neurogenesis.
4.5 Cognitive Domains Affected
- Executive function — planning, judgment, impulse control (prefrontal cortex)
- Working memory and new learning (hippocampus, frontal lobe)
- Processing speed (white matter tracts)
- Visuospatial function and balance (cerebellum)
- Emotional regulation (prefrontal–limbic circuits)
DIAGNOSTIC MARKERS — CNSStructural MRI (grey matter volume, cerebellar atrophy), diffusion tensor imaging (white matter integrity), neuropsychological testing, and clinical gait/cognitive screening (e.g., MoCA).
5. Thiamine Deficiency: Wernicke Encephalopathy and Korsakoff Syndrome
5.1 Wernicke Encephalopathy
An acute neurological emergency from thiamine deficiency, classically presenting with confusion, ataxia, and eye-movement abnormalities — though the full triad appears in only a minority of cases, a major cause of under-diagnosis. Selective neuronal injury occurs in the mammillary bodies, thalamus, and periaqueductal grey matter.
CLINICAL URGENCYWernicke encephalopathy requires immediate high-dose parenteral thiamine, given before glucose in anyone with suspected chronic heavy alcohol use — glucose alone can precipitate or worsen the syndrome. Delay converts reversible acute encephalopathy into permanent brain injury.
5.2 Korsakoff Syndrome
Untreated Wernicke encephalopathy frequently progresses to Korsakoff syndrome — severe anterograde and retrograde amnesia with confabulation (unconscious fabrication, not deliberate lying). Damage centres on the mammillary bodies and thalamic nuclei of the Papez memory circuit. Unlike the acute Wernicke phase, established Korsakoff memory deficits are typically permanent.
5.3 Diagnostic Considerations
- Clinical suspicion in anyone with heavy alcohol history presenting with confusion or poor coordination
- MRI may show mammillary body and periaqueductal signal changes acutely
- Blood thiamine / erythrocyte transketolase activity supports diagnosis — treatment should never wait for results
- Neuropsychological testing distinguishes Korsakoff-pattern amnesia from generalised alcohol-related dementia
6. Peripheral Nervous System Damage
6.1 Alcoholic Peripheral Neuropathy
One of the most common causes of peripheral neuropathy in the developed world, affecting an estimated one-quarter to one-half of long-term heavy drinkers, via direct axonal toxicity plus B1/B6/B12/folate deficiencies common in heavy drinkers.
6.2 Clinical Presentation
Length-dependent, sensory-predominant, symmetric “stocking-glove” numbness, tingling, and burning pain starting in the feet. Autonomic fibres can also be affected — resting tachycardia, orthostatic changes, GI motility disturbances.
6.3 Diagnosis and Reversibility
- Nerve conduction studies typically show an axonal pattern
- Screening for B1, B6, B12, folate deficiency is essential and independently treatable
- Early neuropathy can partially improve with abstinence and repletion; long-standing axonal loss is largely permanent
7. Hepatic (Liver) Damage
The liver bears the largest metabolic burden of ethanol clearance and shows the most well-characterised dose-dependent injury trajectory of any organ, progressing through three broadly recognised stages.
7.1 Stage One: Alcoholic Fatty Liver (Steatosis)
Fat accumulation in hepatocytes, often within days to weeks of sustained intake. Typically asymptomatic and fully reversible with abstinence, generally within 4–6 weeks.
7.2 Stage Two: Alcoholic Hepatitis
Active inflammation with hepatocyte ballooning and Mallory-Denk bodies. Ranges from mild subclinical inflammation to a severe, life-threatening syndrome with jaundice, fever, and liver failure — severe cases carry short-term mortality that can exceed 30–50%.
7.3 Stage Three: Fibrosis and Cirrhosis
Repeated injury-repair cycles activate hepatic stellate cells, depositing collagen and progressively replacing normal architecture with scar tissue. Once reorganised into regenerative nodules, the diagnosis is cirrhosis — irreversible with rare exception — disrupting portal blood flow and synthetic/detox function.
COMPLICATIONS OF ADVANCED CIRRHOSISOesophageal/gastric varices with haemorrhage risk; ascites and spontaneous bacterial peritonitis; hepatic encephalopathy; hepatocellular carcinoma; hepatorenal syndrome.
7.4 Diagnostic Markers
| Marker / Test | What It Detects | Notes |
|---|---|---|
| AST / ALT | Hepatocyte injury | AST:ALT > 2:1 suggests alcoholic aetiology |
| GGT | Enzyme induction / cholestasis | Sensitive but non-specific marker of heavy drinking |
| Bilirubin, INR, albumin | Synthetic/excretory function | Core components of severity scoring |
| Ultrasound / FibroScan | Steatosis, fibrosis staging | Non-invasive alternative to biopsy |
| Liver biopsy | Histological confirmation | Reference standard when non-invasive tests inconclusive |
8. Cardiovascular System
8.1 Alcoholic Cardiomyopathy
Chronic heavy use is one of the most common identifiable causes of dilated cardiomyopathy. Ethanol and acetaldehyde impair contractile protein function and mitochondrial energy production, while oxidative stress and inflammation drive myocardial fibrosis, producing a dilated, poorly contractile left ventricle.
8.2 Arrhythmia — “Holiday Heart Syndrome”
Acute binge consumption is strongly associated with new-onset atrial fibrillation, classically after weekend/holiday binge episodes, via autonomic imbalance, direct electrophysiological effects, and electrolyte disturbances.
8.3 Hypertension
A well-established, dose-dependent relationship with elevated blood pressure, holding even at moderate intake and reversing partially with reduced consumption.
8.4 The “J-Curve” Controversy
Older observational studies suggested a protective effect of light-to-moderate drinking. Recent Mendelian randomisation studies — reducing confounding — largely fail to replicate this, showing risk rising from very low intake levels, suggesting earlier apparent benefit was driven by confounding (e.g. abstainer groups including people who stopped drinking due to illness).
DIAGNOSTIC MARKERS — CARDIOVASCULAREchocardiography (LV dilation, ejection fraction); ECG/ambulatory monitoring (arrhythmia); cardiac MRI (fibrosis); serial blood pressure tracking.
9. Gastrointestinal System and Pancreas
9.1 Gastric and Oesophageal Effects
Ethanol is directly irritant to gastric mucosa, disrupting the protective mucus layer and stimulating acid secretion — causing erosive gastritis and worsening peptic ulcer disease. Vomiting can cause Mallory-Weiss tears or, rarely, Boerhaave syndrome (oesophageal rupture).
9.2 Intestinal Permeability and the Gut–Liver–Brain Axis
Chronic exposure disrupts intestinal tight junctions, increasing gut permeability and allowing bacterial endotoxin translocation — driving hepatic inflammation and, via systemic circulation, neuroinflammation. This axis unifies GI, hepatic, and neurological injury.
9.3 Acute and Chronic Pancreatitis
Alcohol is one of the two leading causes of acute pancreatitis worldwide. Recurrent episodes progress to chronic pancreatitis — irreversible fibrosis, exocrine insufficiency (malabsorption), and a distinct, often difficult-to-manage endocrine insufficiency (diabetes).
9.4 Nutrient Malabsorption
- Thiamine, B6, B12, folate deficiency from reduced intake and impaired absorption
- Zinc and magnesium deficiency, worsening neurological and cardiac function
- Fat-soluble vitamin (A, D, E, K) malabsorption with pancreatic or cholestatic disease
10. Immune System Suppression
Alcohol has complex, dose-dependent immune effects: acute intoxication transiently increases some inflammatory markers, while chronic heavy use produces broad immunosuppression combined with paradoxical chronic low-grade inflammation.
10.1 Innate Immunity
Chronic use impairs neutrophil recruitment and phagocytic function and disrupts mucociliary clearance, explaining increased susceptibility to bacterial pneumonia and tuberculosis reactivation.
10.2 Adaptive Immunity
Reduces T-cell numbers and impairs T-cell/B-cell function, reducing vaccination effectiveness and antibody response.
10.3 Chronic Inflammation
Alongside this immunosuppression, gut-permeability and direct inflammatory signalling in the liver, brain, and vasculature drive sustained low-grade systemic inflammation — damaging tissue while leaving the body less able to fight genuine infection.
11. Endocrine and Metabolic Effects
11.1 Glucose Regulation
Ethanol suppresses hepatic gluconeogenesis, risking significant hypoglycaemia in fasting states. Chronic use is also linked to insulin resistance, type 2 diabetes, and — where chronic pancreatitis damages endocrine tissue — a brittle pancreatogenic diabetes.
11.2 Hypothalamic-Pituitary-Gonadal Axis
Suppresses GnRH pulsatility. In men, combined with testicular Leydig cell toxicity and increased testosterone-to-oestrogen conversion in a damaged liver, this produces hypogonadism: reduced testosterone, gynaecomastia, testicular atrophy, reduced fertility. In women, associated with menstrual irregularity and reduced fertility.
11.3 Adrenal and Stress Axis
Dysregulates the HPA axis, producing pseudo-Cushing syndrome — a state mimicking true Cushing syndrome clinically and biochemically but typically resolving with abstinence.
11.4 Bone-Relevant Endocrine Effects
Suppresses osteoblast activity and disrupts vitamin D metabolism through hepatic dysfunction, feeding directly into the skeletal effects in Section 12.
12. Musculoskeletal System
12.1 Alcoholic Skeletal Myopathy
A dose-dependent proximal myopathy from impaired protein synthesis, mitochondrial dysfunction, and membrane disruption — progressive weakness in hip and shoulder girdle muscles, distinct from acute alcoholic rhabdomyolysis (which can precipitate acute kidney injury).
12.2 Osteoporosis and Fracture Risk
An independent risk factor for reduced bone mineral density via suppressed osteoblast activity and impaired calcium/vitamin D metabolism. Combined with fall risk from neurological impairment (Sections 4 and 6), fracture risk compounds substantially.
13. Reproductive System and Fetal Alcohol Spectrum Disorders
13.2 Fetal Alcohol Spectrum Disorders (FASD)
Ethanol crosses the placenta freely, and the developing fetal brain is exceptionally vulnerable — fetal alcohol dehydrogenase activity is low, prolonging exposure. Alcohol disrupts neuronal migration, synaptogenesis, and myelination, and no gestational stage or quantity has been established as unequivocally safe.
FASD ranges from full fetal alcohol syndrome (characteristic facial features, growth restriction, CNS abnormalities) to milder presentations with cognitive, behavioural, and executive-function deficits without the full facial phenotype — these milder forms are more common and frequently under-diagnosed.
PUBLIC HEALTH CONSENSUSMajor health authorities, including the CDC and equivalent international bodies, advise there is no known safe amount, safe time, or safe type of alcohol during pregnancy or when planning pregnancy.
14. Cancer Risk Across Organ Systems
Ethanol and its metabolite acetaldehyde are classified as a Group 1 carcinogen by IARC — the same category as tobacco smoke and asbestos, reflecting sufficient evidence of causation rather than equivalent potency. Mechanisms include DNA adduct formation, reactive oxygen species, impaired DNA repair, altered folate metabolism, and increased oestrogen relevant to breast cancer.
| Cancer Site | Relationship to Alcohol |
|---|---|
| Oral cavity and pharynx | Strong dose-response; synergistic with tobacco |
| Oesophagus (squamous cell) | Strong association; markedly elevated in ALDH2-deficient individuals |
| Liver (hepatocellular carcinoma) | Arises predominantly in established cirrhosis |
| Colorectum | Consistent moderate dose-response |
| Breast (female) | Risk rises from low intake, partly via oestrogen pathway |
| Larynx | Dose-dependent, synergistic with tobacco |
A critical finding across this literature is the absence of a demonstrated lower threshold for cancer risk — risk appears to rise from the first regular drink, now reflected in updated guidance from several national health authorities.
15. Diagnostic Framework — Clinical and Laboratory Markers
15.1 Consumption Markers vs. Damage Markers
Carbohydrate-deficient transferrin (CDT) and elevated GGT indicate heavy recent consumption but not confirmed structural injury. AST, ALT, bilirubin, and imaging indicate actual damage. This distinction matters because a consumption marker can be abnormal before organ damage occurs — a window for intervention.
| System | Consumption Marker | Damage / Structural Marker |
|---|---|---|
| Liver | GGT, CDT | AST/ALT ratio, bilirubin, INR, elastography, biopsy |
| Brain (structural) | History, cognitive screening | MRI volumetrics, DTI, neuropsychological testing |
| Peripheral nerves | Symptom history | Nerve conduction studies |
| Heart | History, ECG rhythm strip | Echocardiography, cardiac MRI |
| Pancreas | History | Lipase/amylase, faecal elastase |
| Nutritional status | Dietary history | Thiamine, B12, folate, zinc, magnesium |
15.2 Screening Instruments
Structured tools identify at-risk individuals before biomarkers or imaging are indicated. AUDIT and AUDIT-C are the most widely validated internationally; CAGE remains in common clinical use as a rapid screen, though less sensitive to early or lower-severity patterns.
16. Staging and Diagnostic Criteria for Alcohol Use Disorder
The clinical diagnosis framing all organ-specific damage above is Alcohol Use Disorder (AUD), defined in DSM-5 via eleven criteria over a 12-month period, spanning impaired control, social impairment, risky use, and pharmacological indicators (tolerance and withdrawal).
| Severity | Criteria Met (of 11) |
|---|---|
| Mild | 2–3 criteria |
| Moderate | 4–5 criteria |
| Severe | 6 or more criteria |
16.2 Representative Diagnostic Criteria
- Drinking larger amounts or over a longer period than intended
- Persistent desire or unsuccessful efforts to cut down or control use
- Craving, or a strong desire or urge to drink
- Continued use despite awareness of resulting physical or psychological problems
- Tolerance — needing markedly increased amounts for the desired effect
- Withdrawal — a characteristic syndrome on cessation, or drinking to relieve it
Organ damage severity correlates broadly, though imperfectly, with AUD severity and cumulative exposure — the two staging systems are best understood as two views of the same underlying disease process.
17. Reversibility, Recovery Timelines, and Prognosis
A central theme across every organ system is that alcohol-related damage exists on a spectrum from fully reversible to permanently structural, determined chiefly by the point at which exposure stops relative to the transition from functional adaptation to structural cell loss.
| System | Reversible with Abstinence | Typically Irreversible |
|---|---|---|
| Liver | Fatty liver, early fibrosis | Established cirrhosis |
| Brain — grey matter | Partial volume recovery over months | Cerebellar Purkinje cell loss; established Korsakoff amnesia |
| Brain — cognition | Partial to substantial improvement | Long-standing executive/memory deficits may persist |
| Peripheral nerves | Early axonal dysfunction, with repletion | Advanced axonal loss |
| Heart | Early cardiomyopathy, partial-to-full EF recovery | Advanced fibrotic cardiomyopathy |
| Pancreas | Acute pancreatitis (full recovery possible) | Chronic pancreatitis fibrosis |
17.1 Why Timing Matters
Across nearly every system, biology follows a consistent pattern: functional/metabolic adaptation (reversible) followed by structural damage (irreversible). This is why early detection carries disproportionate clinical value relative to detection after structural damage has occurred.
17.2 Neuroplasticity and Partial Brain Recovery
Longitudinal imaging of people with sustained abstinence shows measurable increases in grey matter volume and white matter integrity within months, from reduced neuroinflammation, partial remyelination, and restored neurogenesis. Recovery is most pronounced in cortical grey matter, least pronounced where frank cell loss has occurred (e.g., cerebellar vermis in advanced cases).
18. Conclusion
Alcohol’s damage to the human body follows a small number of shared mechanistic pathways — oxidative stress, mitochondrial dysfunction, chronic inflammation, cofactor depletion, and direct DNA damage from acetaldehyde — that manifest differently depending on the metabolic demands and regenerative capacity of each organ. Neurons and hepatocytes sit at the more vulnerable end of this spectrum precisely because of their high metabolic demand and, for most neurons, their inability to be replaced once lost.
The unifying clinical lesson across every organ system reviewed is consistent: the interval between early, reversible dysfunction and permanent structural damage is the single most important variable in prognosis — which is why screening, early biomarker detection, and timely abstinence remain the most effective interventions available at any stage short of established end-organ disease.
A NOTE ON THIS DOCUMENTThis article is an educational and scientific reference produced for the Makoti Millennium Services Educational Series. It synthesises peer-reviewed literature into a diagnostic framework and is not a substitute for individualised medical assessment. Readers concerned about their own alcohol use are encouraged to consult a licensed physician or addiction medicine specialist. 📋 Copy Full Article Text
Select References and Further Reading
This review synthesises findings from major bodies including the World Health Organization, the U.S. National Institute on Alcohol Abuse and Alcoholism (NIAAA), the International Agency for Research on Cancer (IARC), the U.S. CDC, and peer-reviewed literature in hepatology, neurology, cardiology, and addiction medicine.
- NIAAA Core Resource on Alcohol — mechanisms of organ-specific injury and clinical guidance
- IARC Monographs on the Evaluation of Carcinogenic Risks to Humans — alcohol consumption and ethyl carbamate
- WHO Global Status Report on Alcohol and Health — epidemiological and public health data
- UK Biobank neuroimaging studies on alcohol consumption and grey matter volume
- AASLD — practice guidance on alcohol-associated liver disease
- DSM-5-TR (American Psychiatric Association) — diagnostic criteria for Alcohol Use Disorder





Be First to Comment