
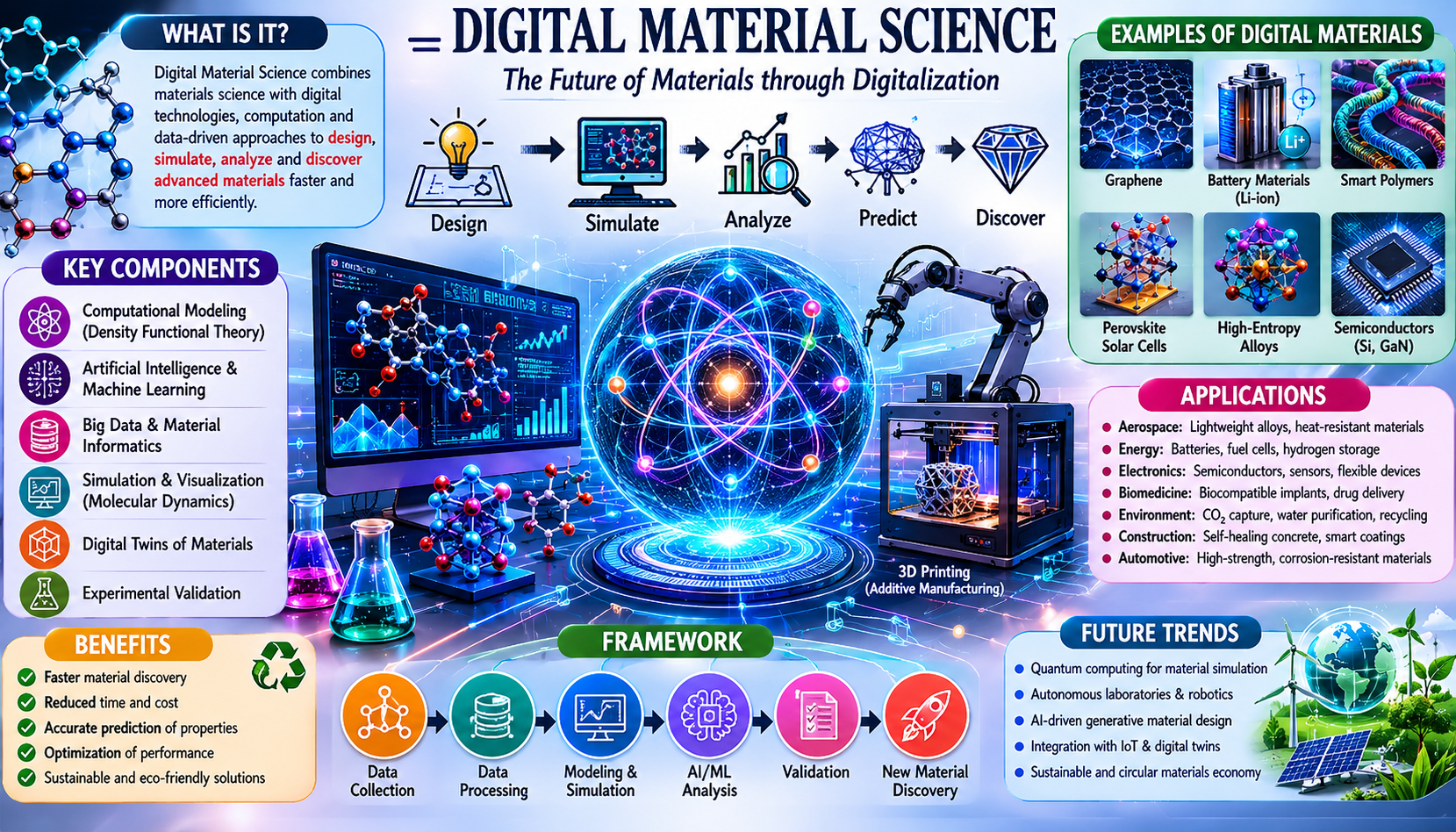
1. Introduction to Digital Material Science
Materials are the foundation of civilization. Every technological leap in human history — from the Bronze Age to the Silicon Revolution — has been defined by humanity’s ability to discover, understand, and harness new materials. Steel enabled skyscrapers. Silicon enabled computers. Carbon fiber enables aerospace. Lithium enabled portable energy storage.
For millennia, materials discovery was empirical: experiment, observe, refine. A blacksmith learned through years of practice. A metallurgist tested thousands of alloys. Even twentieth-century materials science, despite its rigorous theoretical underpinnings, remained largely experimental. The cycle from discovery to deployment stretched over decades.
Digital Material Science fundamentally disrupts this cycle.
At its essence, Digital Material Science is the use of computational tools, mathematical models, data analytics, and artificial intelligence to predict, simulate, design, and optimize the structure, properties, and behavior of materials — without requiring initial physical experimentation. It transforms the materials discovery pipeline from a slow, serendipitous process into a rational, accelerated, data-driven endeavor.
What Makes It “Digital”?
The “digital” in Digital Material Science refers not merely to the use of computers, but to the comprehensive digitization of the materials knowledge pipeline:
- Digital Representation: Materials are represented as mathematical and computational objects — lattice structures encoded in matrices, electron densities as probabilistic fields, mechanical properties as tensors stored in databases.
- Digital Simulation: Physical and chemical behavior is replicated through algorithms — density functional theory, molecular dynamics, finite element analysis — that solve governing equations numerically.
- Digital Discovery: Machine learning and artificial intelligence mine vast material datasets to identify structure-property relationships, predict novel compounds, and recommend synthesis routes.
- Digital Integration: All stages — from atomic structure to engineering application — are connected in seamless computational workflows.
The discipline draws from quantum mechanics, thermodynamics, continuum mechanics, information theory, computer science, and data science. It is inherently interdisciplinary and inherently digital.
2. Historical Context and the Rise of Computational Materials Research
2.1 Early Computational Efforts (1950s–1980s)
The roots of Digital Material Science can be traced to the 1950s, when early computers were first applied to solve quantum mechanical equations for simple atomic systems. Walter Kohn and colleagues laid the groundwork for Density Functional Theory (DFT) in the 1960s — a method that would become one of the most cited scientific approaches in history. DFT allowed researchers to calculate the electronic structure of materials from first principles using only the positions of atomic nuclei as inputs.
In the 1970s and 1980s, the advent of Molecular Dynamics (MD) simulations — pioneered by Alder, Wainwright, and later Rahman — allowed scientists to simulate the motion of atoms over time, governed by classical or semi-classical force fields. These early simulations were limited by computing power: a simulation of a few hundred atoms was computationally demanding.
2.2 The Informatics Revolution (1990s–2000s)
The 1990s brought a qualitative change. The internet enabled global sharing of computational results and experimental data. The Cambridge Structural Database (CSD), the Inorganic Crystal Structure Database (ICSD), and the Materials Project (launched in 2011) began accumulating materials data at unprecedented scale. Researchers recognized that patterns lurking in this data could accelerate discovery far beyond individual experiments or simulations.
The term Materials Informatics emerged to describe the application of data science techniques to materials problems. Early efforts used statistical regression and classical machine learning to correlate compositions with properties such as band gap, melting point, or hardness.
2.3 The AI and Quantum Era (2010s–Present)
The 2010s saw the explosive integration of deep learning into materials science. Neural networks trained on DFT datasets could predict formation energies and band structures in milliseconds — speeds millions of times faster than the underlying quantum calculations. Google DeepMind’s AlphaFold and its materials science analog, GNoME (Graph Networks for Materials Exploration), demonstrated that AI could identify hundreds of thousands of new stable crystalline structures.
Simultaneously, the emergence of quantum computing offered the tantalizing prospect of simulating quantum systems — molecules and materials — with a precision that classical computers fundamentally cannot achieve for large systems. IBM, Google, IonQ, and Quantinuum began developing quantum hardware with direct applications to materials simulation.
The Materials Genome Initiative (MGI), launched by the US government in 2011, institutionalized the digital approach by committing to cut the time and cost of materials discovery in half through computational tools, databases, and data-sharing infrastructure.
3. Thesis: The Digital Twin Paradigm and the Future of Materials
Central Thesis
Digital Material Science constitutes a fundamental paradigm shift in how humanity discovers, develops, and deploys materials. By establishing computational “digital twins” of material systems — representations that exist in silico but faithfully capture physical reality — the discipline collapses the traditional discovery-to-deployment timeline from decades to years or months, democratizes access to advanced materials capabilities, and enables the rational design of materials with properties specifically engineered for defined applications. This paradigm does not replace experimental science; rather, it reconfigures the relationship between theory, computation, and experiment into an iterative, accelerated feedback loop.
Unpacking the Thesis
The concept of the digital twin — a virtual replica of a physical object, system, or process — is central to DMS. In the context of materials:
- A digital twin of a steel alloy might encode its crystal structure, grain boundaries, dislocation densities, and thermo-mechanical properties. It can be “tested” computationally under simulated stress, temperature, and corrosive environments to predict failure modes before a single physical sample is manufactured.
- A digital twin of a battery cathode material might simulate lithium-ion diffusion pathways, charge-discharge cycles, and capacity degradation — enabling the optimization of composition and microstructure for maximum energy density and longevity.
The thesis further argues that this paradigm democratizes materials science. Historically, advanced materials research required costly experimental apparatus accessible only to well-funded institutions. Computational tools — increasingly available through cloud platforms like the Materials Project, AFLOW, and NOMAD — lower barriers, enabling researchers in emerging economies and smaller institutions to participate in frontier materials discovery.
Finally, the thesis identifies a critical distinction: DMS does not eliminate experiment. Rather, it curates experiment. Instead of testing thousands of candidates in the laboratory, a DMS workflow might computationally screen one million candidates, identify the top 50 most promising, and direct experimentalists to synthesize and validate only those. The experimental effort remains essential but is rendered exponentially more efficient.
4. The Foundational Framework of Digital Material Science
Digital Material Science operates through a layered, interconnected framework. We can conceptualize this as a pyramid with five levels:
┌─────────────────────────────┐
│ APPLICATION & DEPLOYMENT │ ← Level 5
└─────────────────────────────┘
┌───────────────────────────────────┐
│ OPTIMIZATION & AI-DRIVEN DESIGN │ ← Level 4
└───────────────────────────────────┘
┌─────────────────────────────────────────┐
│ MULTISCALE INTEGRATION MODELS │ ← Level 3
└─────────────────────────────────────────┘
┌───────────────────────────────────────────────┐
│ SIMULATION ENGINES & DATABASES │ ← Level 2
└───────────────────────────────────────────────┘
┌─────────────────────────────────────────────────────┐
│ QUANTUM MECHANICAL FOUNDATIONS │ ← Level 1
└─────────────────────────────────────────────────────┘Level 1 — Quantum Mechanical Foundations: The bedrock of DMS is quantum mechanics. Properties of materials emerge from the behavior of electrons and nuclei, governed by the Schrödinger equation. DFT and other ab initio methods solve simplified versions of this equation to determine electronic structure from first principles.
Level 2 — Simulation Engines & Databases: At this level, the quantum-derived knowledge is used to run large-scale simulations (Molecular Dynamics, Monte Carlo methods, Phase Field modeling) and to populate materials databases with computed properties.
Level 3 — Multiscale Integration: Materials behavior spans from the angstrom scale (atomic bonds) to the meter scale (structural components). Multiscale models connect these scales, passing information upward — from quantum to atomistic to mesoscale to continuum — to simulate real engineering behavior.
Level 4 — AI-Driven Optimization: Machine learning models trained on simulation and experimental data identify patterns and accelerate prediction. Generative models propose new material compositions. Inverse design algorithms work backward from desired properties to required structures.
Level 5 — Application and Deployment: The validated digital predictions guide physical synthesis and manufacturing. The digital twin continues to function during a material’s operational life, monitoring performance and predicting maintenance needs.
5. Core Theoretical Pillars
5.1 Density Functional Theory (DFT)
DFT is the cornerstone of modern computational materials science. Rather than solving the full many-body Schrödinger equation (intractable for systems with more than a few electrons), DFT reformulates the problem in terms of electron density — a function of only three spatial variables regardless of the number of electrons.
The Hohenberg-Kohn theorems establish that the ground-state energy of a system is a unique functional of its electron density, and the Kohn-Sham equations provide a tractable computational scheme. In practice, DFT can calculate:
- Crystal structure and lattice parameters
- Formation energy and thermodynamic stability
- Electronic band structure (conductor vs. semiconductor vs. insulator)
- Magnetic properties
- Elastic constants and mechanical moduli
- Vibrational properties (phonons) and thermal conductivity
Example: Computing the band gap of gallium nitride (GaN) — used in LEDs — through DFT allows researchers to predict its optical emission wavelength before growing a single crystal. DFT calculations predicted that wurtzite GaN has a direct band gap of ~3.4 eV, consistent with blue-light emission, which was experimentally confirmed and led to the Nobel Prize-winning development of blue LEDs.
5.2 Molecular Dynamics (MD)
MD simulations model materials as collections of atoms interacting through defined force fields. Newton’s equations of motion are integrated numerically over femtosecond timesteps to track atomic trajectories. From these trajectories, thermodynamic, structural, and transport properties are calculated.
Key parameters include:
- Force Field: Mathematical description of atomic interactions (Lennard-Jones, Embedded Atom Method, ReaxFF)
- Ensemble: NPT (constant pressure and temperature), NVT (constant volume and temperature), NVE (microcanonical)
- Timescale: Typically nanoseconds to microseconds for classical MD
Example: MD simulations of water-graphene interfaces have revealed how graphene’s surface energy governs water droplet contact angles — critical information for designing hydrophobic coatings and membrane filtration systems.
5.3 Phase Field Modeling
Phase field models describe the microstructural evolution of materials — grain growth, solidification, precipitate formation, crack propagation — using continuous order parameter fields. The Allen-Cahn and Cahn-Hilliard equations govern the time evolution of these fields, allowing mesoscale simulation without explicit tracking of sharp interfaces.
Example: Phase field modeling of nickel superalloy solidification predicts the dendritic microstructure that forms during casting, enabling optimization of cooling rates to minimize defects in turbine blades.
5.4 Finite Element Analysis (FEA)
At the continuum scale, FEA discretizes complex geometries into finite elements and solves governing partial differential equations (stress-strain relations, heat conduction, fluid flow) numerically. In DMS, FEA is often fed with material properties derived from lower-scale simulations.
Example: A digital twin of a carbon fiber reinforced polymer (CFRP) aircraft fuselage uses FEA to simulate structural loads and predict fatigue life, with fiber orientation and ply properties informed by molecular-scale DFT calculations of fiber-matrix adhesion.
6. Simulation Methodologies
6.1 Ab Initio Methods
Ab initio (Latin: “from the beginning”) methods require no empirical input. The most common is DFT, but higher-accuracy methods include:
- Hartree-Fock (HF): Approximates many-electron wavefunctions as antisymmetric products of single-electron wavefunctions.
- MP2 and Coupled Cluster (CCSD(T)): Post-HF methods that add electron correlation corrections. CCSD(T) is considered the “gold standard” for molecular calculations.
- GW Approximation: Improves on DFT for band gap calculations by accounting for quasiparticle effects.
- TDDFT: Time-dependent DFT for optical properties and excited states.
6.2 Force Field-Based Simulations
When quantum accuracy is unnecessary or computationally prohibitive (for large systems or long timescales), force field-based methods provide a tractable alternative:
- Classical MD: Atoms are point masses; electronic degrees of freedom are implicit in the force field.
- Coarse-Grained MD (CGMD): Groups of atoms are treated as single beads, enabling simulation of polymers and biological membranes at longer timescales.
- ReaxFF: Reactive force fields that allow bond formation and breaking, bridging classical and quantum descriptions.
6.3 Monte Carlo Methods
Monte Carlo (MC) methods use random sampling to explore the configuration space of a material system. Unlike MD (which evolves systems in real time), MC generates statistically representative configurations, making it ideal for thermodynamic equilibrium properties.
- Grand Canonical Monte Carlo (GCMC): Simulates adsorption — critical for studying gas storage in porous materials like metal-organic frameworks (MOFs).
- Kinetic Monte Carlo (KMC): Simulates rare events (diffusion, nucleation) over timescales inaccessible to MD.
6.4 CALPHAD (Calculation of Phase Diagrams)
CALPHAD uses thermodynamic databases — built from experimental measurements and DFT calculations — to compute multicomponent phase diagrams. It is indispensable for alloy design, predicting which phases are thermodynamically stable under given conditions.
Example: CALPHAD calculations guided the design of multi-principal-element alloys (also called high-entropy alloys), predicting phase stability in five or more component systems that would be impractical to map experimentally.
7. Machine Learning and AI in Materials Discovery
7.1 The Paradigm Shift
Traditional computational materials science is computationally expensive. A high-quality DFT calculation for a moderately complex system (100–200 atoms) might take hours to days on a supercomputer. Machine learning offers a compelling complement: train models on DFT datasets, then use those models to predict properties orders of magnitude faster.
This approach — training ML models on quantum mechanical data — is called Machine Learning Interatomic Potentials (MLIPs) or ML Force Fields. Pioneering work by Behler and Parrinello (2007) demonstrated that neural networks could represent the potential energy surface of materials with near-DFT accuracy at a fraction of the computational cost.
7.2 Types of ML Models in DMS
Descriptors and Feature Engineering: Early ML models for materials required hand-crafted features — mathematical representations of atomic environments (Coulomb matrix, symmetry functions, SOAP descriptors). These features capture local chemical environments and are fed into regression or neural network models.
Graph Neural Networks (GNNs): Modern approaches represent crystal structures as graphs: atoms are nodes, bonds are edges, and properties of edges encode interatomic distances and angles. GNNs learn representations that are invariant to rotation, reflection, and translation — essential for physical consistency. Architectures such as SchNet, MEGNet, DimeNet, and CGCNN have achieved state-of-the-art accuracy on materials property prediction benchmarks.
Generative Models: Variational autoencoders (VAEs) and generative adversarial networks (GANs) can generate new material compositions and crystal structures by sampling from learned latent spaces. These models enable inverse design: specify desired properties, generate structures predicted to exhibit them.
Reinforcement Learning: RL agents can be trained to navigate the compositional space of materials, proposing and evaluating candidates in a guided search — analogous to how AlphaGo learned to play the game of Go.
7.3 Case Study: AlphaFold and GNoME
AlphaFold (DeepMind, 2020–2021) predicted the three-dimensional structure of virtually every known protein with remarkable accuracy — a problem that had resisted experimental and computational solution for decades. While proteins are biological molecules rather than inorganic materials, AlphaFold’s methodology demonstrated the transformative power of deep learning applied to molecular structure prediction.
GNoME (Graph Networks for Materials Exploration, DeepMind, 2023) extended this philosophy to inorganic crystals. GNoME predicted 2.2 million new stable crystal structures — expanding the known space of stable inorganic compounds by nearly an order of magnitude. Of these, approximately 380,000 were identified as particularly promising for experimental synthesis. Experimental validation confirmed the stability of several predicted structures, representing a landmark demonstration of AI-driven materials discovery.
7.4 Transfer Learning and Limited Data
A persistent challenge in ML for materials is the scarcity of labeled training data. Unlike image recognition (millions of labeled examples), DFT databases may contain tens of thousands to a few hundred thousand entries.
Transfer learning — pre-training on large datasets and fine-tuning on smaller domain-specific datasets — has proven effective. Models pre-trained on comprehensive DFT databases (like the Materials Project’s ~150,000 computed compounds) can be fine-tuned for specific property prediction tasks with minimal additional data.
8. Digital Databases and Informatics
8.1 The Role of Materials Databases
Materials databases are the information infrastructure of DMS. They store computed and experimental data — crystal structures, formation energies, band gaps, elastic properties, magnetic moments — in standardized, queryable formats. The principal public databases include:
- Materials Project (materialsproject.org): Over 150,000 inorganic compounds with DFT-computed properties. Developed at Lawrence Berkeley National Laboratory.
- AFLOW (aflow.org): Over 3 million entries computed with standardized DFT workflows. Emphasizes high-throughput alloy and compound screening.
- NOMAD (nomad-lab.eu): Repository of raw computational data — input and output files from DFT codes — enabling reproducibility and AI training.
- ICSD (Inorganic Crystal Structure Database): Experimental crystal structure data for over 250,000 compounds.
- OQMD (Open Quantum Materials Database): Thermochemical properties computed at scale.
- COD (Crystallography Open Database): Open-access repository of crystal structures from peer-reviewed publications.
8.2 High-Throughput Computational Screening
High-throughput (HT) computing involves the automated, systematic execution of thousands to millions of calculations using standardized computational workflows. HT approaches enabled the screening of metal-organic frameworks (MOFs) for carbon capture, identifying candidates from a space of ~100,000 MOF structures in a search that would have required centuries of experimental effort.
Workflow Management Systems such as AiiDA, FireWorks, and Atomate automate the orchestration of HT calculations, ensuring reproducibility, tracking provenance, and managing the massive data flows generated by large-scale computational campaigns.
8.3 FAIR Data Principles
Materials databases increasingly adhere to the FAIR principles — that data should be:
- Findable: Assigned persistent identifiers and rich metadata
- Accessible: Retrievable through open, standardized protocols
- Interoperable: Using community-agreed formats and ontologies
- Reusable: Released with clear usage licenses and provenance
FAIR principles are essential for enabling AI models to train on diverse materials datasets and for ensuring the long-term reproducibility of computational results.
9. Quantum Computing in Materials Science
9.1 Why Quantum Computers for Materials?
The fundamental challenge of simulating quantum systems on classical computers is exponential scaling. Representing the full quantum state of a system of N electrons requires memory that grows as 2^N — rendering exact simulations impossible for systems larger than ~50 electrons on classical hardware.
Quantum computers, operating on qubits that can exist in superposition and entanglement, are believed to simulate quantum systems efficiently. The quantum simulation algorithms (Variational Quantum Eigensolver, Quantum Phase Estimation) directly exploit quantum mechanics to calculate molecular and material properties.
9.2 Current State and Near-Term Applications
As of the mid-2020s, quantum computers remain in the “noisy intermediate-scale quantum” (NISQ) era — devices with 50–1000 qubits, subject to significant noise and decoherence. Current applications are limited to small molecules (H₂, LiH, N₂ binding energy) and simple model Hamiltonians.
However, near-term demonstrations have shown:
- VQE (Variational Quantum Eigensolver): Calculates ground state energies of small molecules on quantum hardware, agreeing with classical results.
- Quantum simulation of Fermi-Hubbard models: Relevant to high-temperature superconductor physics.
The critical threshold — “quantum advantage” over classical computers for practically relevant materials problems — likely requires fault-tolerant quantum computers with millions of physical qubits, anticipated in the 2030s by leading estimates.
9.3 Hybrid Quantum-Classical Algorithms
Most near-term quantum materials simulations use hybrid approaches: a classical optimization loop drives the parameters of a quantum circuit, combining the strengths of both architectures. The VQE is the archetype of this approach: a quantum circuit prepares trial wavefunctions, a quantum processor evaluates energies, and a classical optimizer adjusts circuit parameters to minimize energy.
10. Multiscale Modeling: From Atoms to Engineering Components
10.1 The Challenge of Scale
Material behavior spans roughly twelve orders of magnitude in length scale:
| Scale | Range | Governing Physics | Simulation Method |
|---|---|---|---|
| Electronic | 0.1–1 Å | Quantum mechanics | DFT, HF, TDDFT |
| Atomic | 1–100 Å | Classical/quantum | MD, MC |
| Mesoscale | 10 nm–10 μm | Statistical mechanics | Phase field, CGMD |
| Microscale | 1–1000 μm | Continuum mechanics | FEA, CFD |
| Macroscale | mm–m | Engineering mechanics | FEA, structural analysis |
No single simulation method spans all these scales. Multiscale modeling develops systematic strategies to bridge them.
10.2 Sequential (Hierarchical) Multiscale Methods
In sequential multiscale approaches, simulations at finer scales generate parameters (force fields, constitutive laws, material properties) that are passed up to coarser-scale models:
- DFT → calculates interatomic potentials → MD uses them
- MD → calculates grain boundary energies → Phase field uses them
- Phase field → calculates microstructure → FEA uses it
This is sometimes called bottom-up or information-passing multiscale modeling.
10.3 Concurrent Multiscale Methods
Concurrent approaches divide a simulation domain into regions handled by different methods simultaneously. The most famous example is the QM/MM (Quantum Mechanics / Molecular Mechanics) method:
- A small chemically active region (e.g., a crack tip, an enzyme active site) is treated quantum mechanically with DFT
- The surrounding region is treated with classical force fields
- A handshaking scheme manages the interface
The ONIOM method (Maseras and Morokuma) formalized this approach and it has become standard in computational chemistry and increasingly in materials science.
10.4 Data-Driven Multiscale Methods
Emerging data-driven approaches use ML to directly learn the constitutive relationships that connect scales, bypassing the need for explicit multiscale coupling schemes. Neural network potentials trained on DFT data can simulate millions of atoms with near-quantum accuracy — effectively achieving quantum mechanical fidelity at an atomistic scale.
11. Real-World Applications and Case Studies
11.1 Battery Materials Design
The global transition to electric vehicles and renewable energy storage is critically dependent on advanced battery materials. DMS has become central to the accelerated development of next-generation batteries.
Cathode Materials: High-throughput DFT screening of lithium transition metal oxides has identified promising cathode compositions beyond conventional LiCoO₂. The AFLOW and Materials Project databases have been queried to identify materials with high theoretical capacity, appropriate voltage, and structural stability. DFT calculations revealed that lithium-rich layered oxides (Li₁.₂Ni₀.₁₃Mn₀.₅₄Co₀.₁₃O₂) could deliver theoretical capacities exceeding 250 mAh/g.
Solid-State Electrolytes: MD simulations have been used to predict lithium-ion conductivity in solid electrolyte candidates — a key property governing battery performance. Screening of garnet-type oxides (Li₇La₃Zr₂O₁₂) using MD correctly predicted the superionic behavior that was subsequently confirmed experimentally.
Case Study — Citroen C5 Battery Optimization: Researchers at the Toyota Research Institute used high-throughput DFT and ML screening to evaluate over 12,000 candidate solid-state electrolyte compositions. The computational screening narrowed the field to 23 promising candidates, of which 5 were subsequently synthesized and characterized. This reduced the experimental workload by over 99.8% relative to an exhaustive experimental search.
11.2 Aerospace Superalloys
Nickel-based superalloys — used in jet turbine blades — must maintain strength and oxidation resistance at temperatures exceeding 1,000°C. Designing new compositions is extraordinarily complex: modern superalloys contain 10 or more elemental components.
DMS approaches for superalloy design integrate:
- CALPHAD phase diagram calculations to predict stable phase assemblages
- MD simulations to evaluate diffusion and creep behavior
- FEA to simulate stress distribution in turbine blade geometries
- ML models trained on historical alloy property databases to predict hot corrosion resistance
Rolls-Royce and GE Aviation have both reported using integrated computational materials engineering (ICME) frameworks — a formal embodiment of DMS principles — to reduce superalloy development timelines from ~20 years to ~7 years.
11.3 Pharmaceutical Solid Forms
While not “traditional” materials science, the design of pharmaceutical solid forms (polymorphs, cocrystals, salts) is a materials problem with enormous societal impact. Drug molecules can crystallize in multiple solid forms with different solubility, stability, and bioavailability profiles.
Crystal structure prediction (CSP) — a DMS methodology — computationally generates and ranks thousands of possible packing arrangements of drug molecules. DFT calculations then refine the energies and determine which polymorphs are thermodynamically stable.
AstraZeneca and Pfizer have integrated CSP into their drug development pipelines, identifying stable polymorphs computationally before manufacturing scale-up, preventing costly post-commercialization polymorph surprises (as occurred historically with ritonavir, the HIV drug that crystallized into a less soluble form after market launch).
11.4 Metal-Organic Frameworks for Carbon Capture
Metal-organic frameworks (MOFs) are porous crystalline materials with extraordinarily high internal surface areas — up to 7,800 m²/g — making them candidates for gas storage, separation, and carbon capture. The combinatorial space of possible MOF compositions (metal nodes, organic linkers, topology) is astronomically large.
High-throughput computational screening — using GCMC simulations and DFT-derived force fields — has been applied to databases of over 500,000 hypothetical MOF structures to identify candidates for selective CO₂ capture. This computational screening identified several structures with CO₂ working capacities and selectivities superior to leading experimental candidates, providing prioritized synthesis targets.
11.5 Semiconductor Design
The semiconductor industry relies extensively on DMS. Band gap engineering — tuning the electronic band gap of semiconductors for specific optoelectronic applications — uses DFT and many-body perturbation theory (GW calculations) to predict band structures of novel compositions.
Perovskite solar cells: DFT calculations on hybrid organic-inorganic perovskites (ABX₃ structures, where A = methylammonium, B = Pb/Sn, X = halide) predicted tunable band gaps across the visible spectrum. This guided the experimental development of perovskite solar cells with efficiencies rising from ~3% in 2009 to over 25% by 2023 — a rise accelerated significantly by computational guidance.
11.6 Biomaterials and Medical Implants
DMS increasingly informs the design of biomaterials for orthopedic implants, dental prosthetics, and drug delivery systems:
- MD simulations of protein adsorption onto titanium surfaces elucidate biocompatibility mechanisms
- FEA models of hip implant stress distribution predict failure sites and guide geometric optimization
- ML models predict the degradation rates of biodegradable polymers (PLA, PLGA) in physiological environments
12. Challenges, Limitations, and Ethical Considerations
12.1 Accuracy vs. Computational Cost
A persistent tension in DMS is the trade-off between accuracy and computational tractability. DFT is approximate — the exchange-correlation functional is an uncontrolled approximation. Standard DFT underestimates band gaps by 30–50%, and notoriously fails to predict the Mott insulating behavior of transition metal oxides. Higher-accuracy methods (GW, CCSD(T)) correct these deficiencies but are far more expensive.
For ML models, accuracy is only as good as the training data. If the training database is biased — over-representing certain chemical spaces — the model will perform poorly outside that space.
12.2 The Validation Gap
A critical challenge is bridging the gap between computational prediction and experimental validation. Computationally “stable” structures may not be experimentally synthesizable due to kinetic barriers. Calculated properties assume ideal single crystals; real materials contain defects, surfaces, grain boundaries, and residual stresses that alter behavior.
The field increasingly addresses this through “synthesizability” metrics — ML models trained to predict whether a computationally stable compound can actually be made in the laboratory — and through the development of computational tools for defect and surface modeling.
12.3 Data Scarcity and Bias
While materials databases have grown enormously, they remain sparse relative to the vast combinatorial space of possible materials. Experimental databases are biased toward materials that have historically been of interest (oxides, metals, semiconductors), leaving large chemical spaces under-characterized.
Active learning approaches address this by intelligently selecting which calculations or experiments to perform next — maximizing information gained per computational dollar spent.
12.4 Reproducibility
Computational reproducibility — a cornerstone of scientific integrity — is an ongoing challenge in DMS. Different DFT codes, pseudopotentials, basis sets, and convergence parameters can yield different results for the same material. Initiatives like the Delta Project have quantified these discrepancies and promoted standardized benchmarking.
The FAIR data principles (Section 8.3) and workflow management systems (AiiDA, FireWorks) directly address reproducibility by capturing complete computational provenance.
12.5 Ethical Considerations
Digital Material Science carries ethical dimensions:
- Dual Use: Computational tools for materials design could accelerate the development of materials for weapons systems, including novel energetic materials or armors, raising questions about access control and publication ethics.
- Environmental Impact of Computing: Large-scale HT screening and ML training consume enormous energy. The carbon footprint of a large DFT computational campaign must be weighed against its scientific benefit.
- Democratization vs. Concentration: While cloud-based DMS tools lower barriers, the most powerful capabilities (large-scale quantum computing, proprietary ML models trained on vast datasets) may concentrate in the hands of a few large corporations, widening the gap between institutions.
- Data Ownership: Questions of intellectual property in materials data — who owns a computationally predicted crystal structure? — remain legally unsettled.
13. The Future Landscape
13.1 Self-Driving Laboratories
The logical culmination of the DMS paradigm is the self-driving laboratory — an autonomous experimental system guided by AI. Robotic platforms synthesize and characterize materials; ML algorithms analyze results in real time; active learning algorithms decide what to try next — forming a closed loop that operates continuously without human intervention.
The Acceleration Consortium (University of Toronto), NIST’s Center for Autonomous Chemistry, and A-Lab at Lawrence Berkeley National Laboratory have demonstrated self-driving laboratories that autonomously synthesize novel inorganic compounds, confirming AI-predicted structures at rates impossible with traditional laboratory practice.
13.2 Foundation Models for Materials
By analogy with large language models (LLMs) like GPT-4 in natural language processing, the materials community is developing foundation models — large neural networks pre-trained on diverse materials datasets that can be fine-tuned for specific downstream tasks. Models such as MACE-MP-0 and SevenNet have demonstrated that a single pre-trained model can serve as a universal interatomic potential across the periodic table, enabling MD simulations of virtually any material without bespoke force field development.
13.3 Digital-Experimental Convergence
The distinction between digital and experimental materials science will continue to blur. Experiments increasingly generate digital data in real time — synchrotron X-ray measurements, electron microscopy images, spectroscopic data — that directly feed ML models. Digital simulations increasingly direct experimental synthesis routes. The future laboratory is a hybrid cyber-physical environment.
13.4 Quantum Computing Maturation
As quantum hardware advances toward fault tolerance, quantum simulation of materials will become a practical tool for problems intractable classically. Electronic structure calculations for strongly correlated materials (high-temperature superconductors, catalysts with complex active sites) are the most anticipated near-term quantum advantage applications.
13.5 Sustainability-Driven Materials Design
Climate change mitigation drives urgent materials challenges: lightweight structural materials for transportation, efficient photovoltaics, electrocatalysts for hydrogen production, materials for carbon capture and utilization. DMS provides the rapid screening capability needed to meet these challenges on timescales commensurate with the climate crisis.
14. Glossary of Key Terms
Ab Initio: Latin for “from the beginning”; refers to computational methods that derive results solely from fundamental physical principles, without empirical input.
Band Gap: The energy difference between the top of the valence band and the bottom of the conduction band in a semiconductor or insulator; determines electrical and optical behavior.
CALPHAD: Calculation of Phase Diagrams; a thermodynamic modeling approach using databases of Gibbs energy functions to predict phase stability in multicomponent systems.
Crystal Structure: The three-dimensional periodic arrangement of atoms in a crystalline material, defined by a unit cell and space group symmetry.
Density Functional Theory (DFT): A quantum mechanical method for computing the electronic structure of atoms, molecules, and solids, based on representing the quantum state in terms of electron density rather than the many-body wavefunction.
Digital Twin: A virtual model that replicates the structure, properties, and behavior of a physical material or system, enabling computational prediction and monitoring.
Force Field: An empirical mathematical function describing interatomic interactions, used in classical molecular dynamics and Monte Carlo simulations.
High-Entropy Alloy (HEA): An alloy containing five or more principal elements in near-equimolar concentrations; often exhibits unusual combinations of strength, ductility, and corrosion resistance.
Integrated Computational Materials Engineering (ICME): An engineering approach that integrates computational tools across multiple scales — from quantum to continuum — to accelerate materials development and qualification.
Machine Learning Interatomic Potential (MLIP): A potential energy surface represented by a machine learning model (typically a neural network) trained on quantum mechanical data.
Materials Genome Initiative (MGI): A US government initiative launched in 2011 to double the speed of materials discovery and deployment through computational tools, data sharing, and workforce development.
Metal-Organic Framework (MOF): A class of porous crystalline materials formed by coordination of metal ions or clusters with organic linker molecules; characterized by high surface area and tunable pore geometry.
Molecular Dynamics (MD): A simulation method in which the time evolution of a system of atoms is computed by numerically integrating Newton’s equations of motion.
Monte Carlo (MC): A computational method using random sampling to evaluate statistical mechanical properties of materials systems.
Perovskite: A class of materials with the ABX₃ crystal structure; encompasses a wide range of functional materials including ferroelectrics, superconductors, and photovoltaics.
Phase Field: A computational method for simulating microstructural evolution using continuous order parameter fields that track the spatial distribution of phases.
QM/MM: Quantum Mechanics / Molecular Mechanics; a hybrid simulation method treating chemically active regions quantum mechanically and surrounding regions with classical force fields.
Quasiparticle: An emergent excitation in a many-body quantum system that behaves as an effective particle; electrons in solids behave as quasiparticles with properties modified by interactions.
Variational Quantum Eigensolver (VQE): A hybrid quantum-classical algorithm for computing the ground state energy of quantum systems using a quantum computer.
15. References and Further Reading
Foundational Papers
- Hohenberg, P. & Kohn, W. (1964). Inhomogeneous Electron Gas. Physical Review, 136(3B), B864.
- Kohn, W. & Sham, L.J. (1965). Self-Consistent Equations Including Exchange and Correlation Effects. Physical Review, 140(4A), A1133.
- Behler, J. & Parrinello, M. (2007). Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Physical Review Letters, 98(14), 146401.
- Curtarolo, S. et al. (2012). The high-throughput highway to computational materials design. Nature Materials, 11, 191–202.
- Jain, A. et al. (2013). Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Materials, 1(1), 011002.
Key Review Articles
- de Pablo, J.J. et al. (2019). New frontiers for the materials genome initiative. npj Computational Materials, 5, 41.
- Schmidt, J. et al. (2019). Recent advances and applications of machine learning in solid-state materials science. npj Computational Materials, 5, 83.
- Noé, F. et al. (2020). Machine learning for molecular simulation. Annual Review of Physical Chemistry, 71, 361–390.
- Merchant, A. et al. (2023). Scaling deep learning for materials discovery. Nature, 624, 80–85. [GNoME paper]
Books
- Martin, R.M. (2020). Electronic Structure: Basic Theory and Practical Methods (2nd ed.). Cambridge University Press.
- Frenkel, D. & Smit, B. (2023). Understanding Molecular Simulation: From Algorithms to Applications (3rd ed.). Academic Press.
- Rajan, K. (2013). Informatics for Materials Science and Engineering. Butterworth-Heinemann.
- Tadmor, E.B. & Miller, R.E. (2011). Modeling Materials: Continuum, Atomistic and Multiscale Techniques. Cambridge University Press.
Online Resources and Databases
- Materials Project: https://materialsproject.org
- AFLOW: http://aflow.org
- NOMAD Repository: https://nomad-lab.eu
- ICSD (Inorganic Crystal Structure Database): https://icsd.fiz-karlsruhe.de
- AiiDA Workflow Manager: https://www.aiida.net
- VESTA Crystal Structure Visualization: https://jp-minerals.org/vesta
Conclusion
Digital Material Science has matured from a niche computational tool into the central engine of materials innovation. It unifies quantum mechanics, statistical physics, computational chemistry, materials informatics, and artificial intelligence into a coherent, accelerated discovery pipeline. The evidence reviewed across this article supports the central thesis: the digital twin paradigm is not a supplement to experimental materials science, but its new organizing principle.
From battery materials that will power the electric economy to superalloys enabling cleaner aviation; from pharmaceutical crystal design to metal-organic frameworks capturing atmospheric carbon — DMS is actively reshaping every domain of applied materials research. The self-driving laboratory, the universal foundation model, and the fault-tolerant quantum computer are not distant speculations but near-horizon realities.
The materials scientist of the coming decades will be fluent in both the language of atoms and the language of data. They will operate at the boundary of the physical and the digital, translating between the two with the confidence that only a deep, integrated understanding of Digital Material Science can provide.
The materials that will define the next century of civilization are, in a very real sense, already taking shape — in equations, in algorithms, in the silent dance of electrons simulated on supercomputing clusters around the world. The laboratory bench remains indispensable, but the first brush stroke is now drawn in code.




Be First to Comment